
行业资料共192篇 第4页
排序

21 CFR Part 312 Investigational New Drug Application (IND)-新药临床试验申请 中英文双语对照
《21 CFR Part 312》是美国食品药品监督管理局(FDA)制定的关于新药临床试验申请 (IND)的管理法规,规定了药物在正式上市前开展临床研究的全过程监管要求。该法规最初于 1987 年发布,...

202510-ECA -Audit Trail Review SOP General Draft审计追踪审核实操指南-双语版下载
数据完整性是产品质量与患者安全的核心保障,而审计追踪(Audit Trail)作为记录数据全生命周期变更的关键工具,其合规审查已成为 GMP(药品生产质量管理规范)监管的核心要点。ECA在10月份发布...

资源分享-中检院-生物制品检验技术操作规范2019版
本书为《中国食品药品检验检测技术系列丛书》之一,其内容主要包括通用检测方法和各类生物制品的特异性检测方法。通用检测方法,只收录了至少两大类制品共同使用的方法,有些方法虽然在某一大类...

资源分享-202512-美国药典凡例(中英文对照)-允咨
该文件为美国药典(USP-NF)凡例的中英文对照版本,核心围绕药典的适用范围、关键规则及具体要求展开,明确其适用于 USP 和 NF 认可的所有专论与通则,规定了药典的标题版本(含常规修订与加速...

海南药监-关于药品变更相关问题的百问百答-附下载
全文下载链接:https://pan.quark.cn/s/edac9bc8417e 第一篇 申报流程 问题 1:药品备案类变更受理形式审查及技术审查流程是什么? 答:1. 申请人提交申请申请人按照《国家药...

资源分享-制药企业有害生物防治管理指南T/CPCACN 0021—2025
文件规定了制药企业有害生物防制原则、各方职责、防制管理、偏差调查、沟通与培训、文件管理、安全管理。文件适用于指引制药企业的有害生物防制管理。链接:https://pan.quark.cn/s/7453d28c3e6...
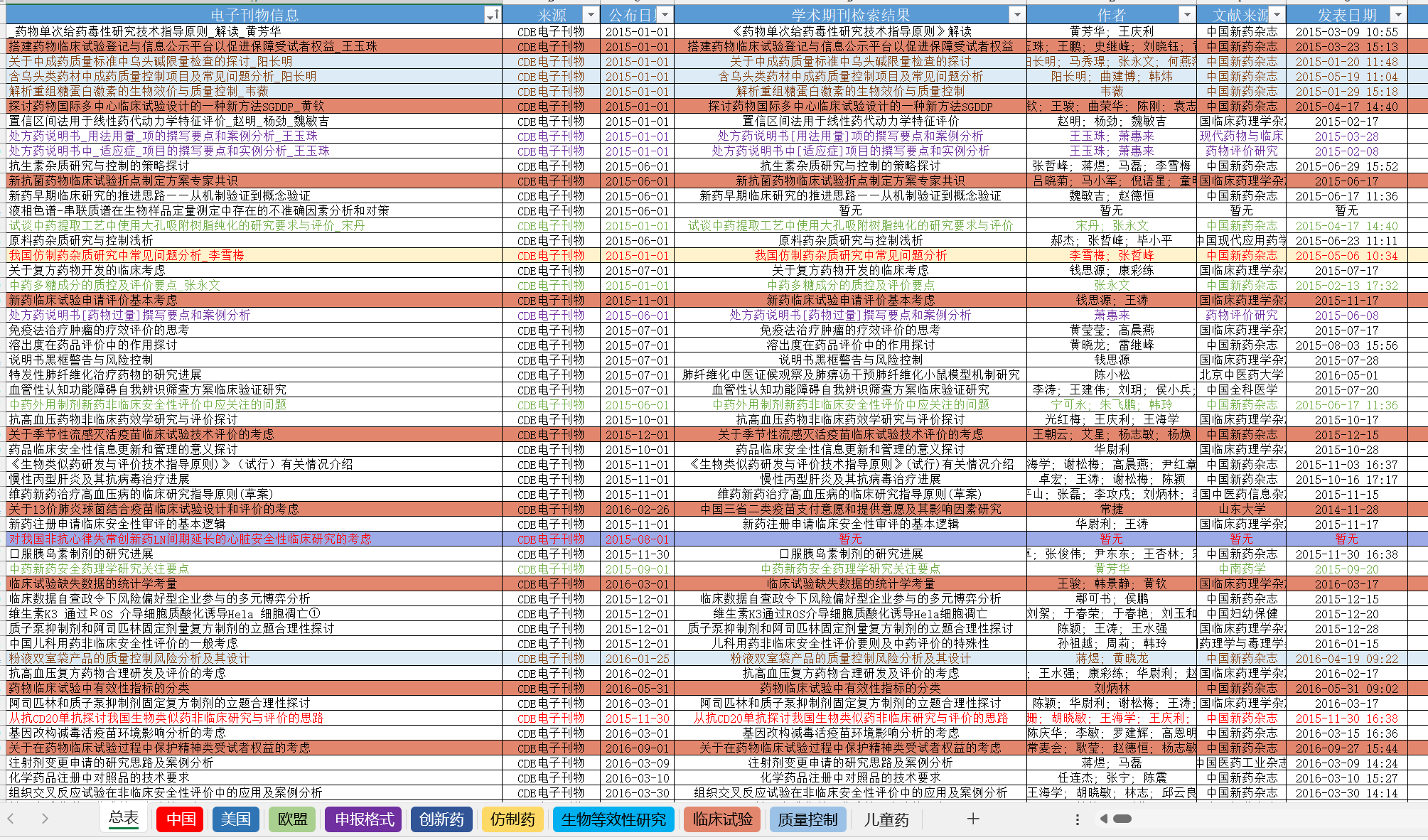
资源分享-CDE电子刊物总览
引言 在药品研发、审评和监管领域,及时获取专业、全面的信息至关重要。今天要给大家分享一份资源 ——CDE 电子刊物总览(2015-至今),这是一个涵盖丰富药品领域知识的信息宝库。 内...

CDE大湾区-多产品共线生产的风险管理-资料分享
在药品生产领域,多产品共线生产是常见现象,但伴随的污染与交叉污染风险不容忽视。药品审评检查大湾区分中心组织了培训,主要包含以下内容 多产品共线生产风险概述 多产品共线生产风险评...

资源分享-CMDE-中国医疗器械器审中心答疑解惑汇总(202511)
医疗器械器审中心发布的 6.3 版答疑解惑汇总(2025 年 11 月 17 日),收录了 2017 年至 2025 年间的 530 个核心问题及解答,聚焦医疗器械与体外诊断试剂注册申报、临床评价、检测验证、技术要...

资源分享-山东药监-药品GMP检查手册(上,下)
本书是专为药品GMP检查员、监管人员和药企从业者设计的便携式工具书。它于2021年2月发布,旨在配合新药品管理法的实施,推动药品生产质量管理水平的提升。 上册(法规篇):基础法律法规...





kidzhangy7月前0
感谢分享,楼主好人一生平安Terrific8月前0
感谢!特别有用,资源分享的很及时!北岛2年前0
相当于货架期多久?Mark4年前2
Thanks for your blog, nice to read. Do not stop.一位WordPress评论者5年前0
嗨,这是一条评论。 要开始审核、编辑及删除评论,请访问仪表盘的“评论”页面。 评论者头像来自Gravatar。