
行业资料共192篇 第3页
排序

资源分享-中检院2014版-化学药品对照品图谱集质谱
谱是用于化合物结构鉴定的主要检测手段之一。从1912年第一台质谱仪诞生以来,质谱技术在样品导入、离子源和分析器方面都取得了巨大的进展。目前,根据离子源不同可分为电子轰击离子化(EI)、化学...

资源分享-CFDI20251015生物制品特殊生产设备检查关注点-康鹰
生物制品作为现代医药领域的核心赛道,其质量直接关系到公众健康与用药安全。而生产设备作为工艺实施的核心载体,是生物制品质量控制的关键环节。国家药监局核查中心(CFDI)针对生物制品生产设...

202510-ECA -Audit Trail Review SOP General Draft审计追踪审核实操指南-双语版下载
数据完整性是产品质量与患者安全的核心保障,而审计追踪(Audit Trail)作为记录数据全生命周期变更的关键工具,其合规审查已成为 GMP(药品生产质量管理规范)监管的核心要点。ECA在10月份发布...

资源分享 20251212-苏州无菌大会ppt
2025 年 12 月 11 日 - 12 日在苏州尹山湖希尔顿酒店举办的第一届无菌药品质量安全大会相关资料,聚焦全球监管标准融合与质量体系升级背景下无菌药品质量安全,围绕 2025 版《无菌药品附录》《...

IT基础设施控制与合规性:制药行业GxP指南解析
今天,我想和大家分享一份关于IT基础设施控制和合规性的笔记文档分析。这份文档名为202512《IT基础设施控制和符合性笔记》,总共48页,主要针对制药行业的GxP法规环境,提供了一种结构化的方法...

PDA-TR56-阶段适宜性质量体系与药品生产质量管理规范在生物药原液研发中的应用2026修订版-附下载
近日,Parenteral Drug Association(PDA)正式推出《PDA 技术报告第 56 号(修订版):阶段适宜性质量体系与药品生产质量管理规范在生物药原液研发中的应用》(以下简称 TR56 2026 修订版)。...
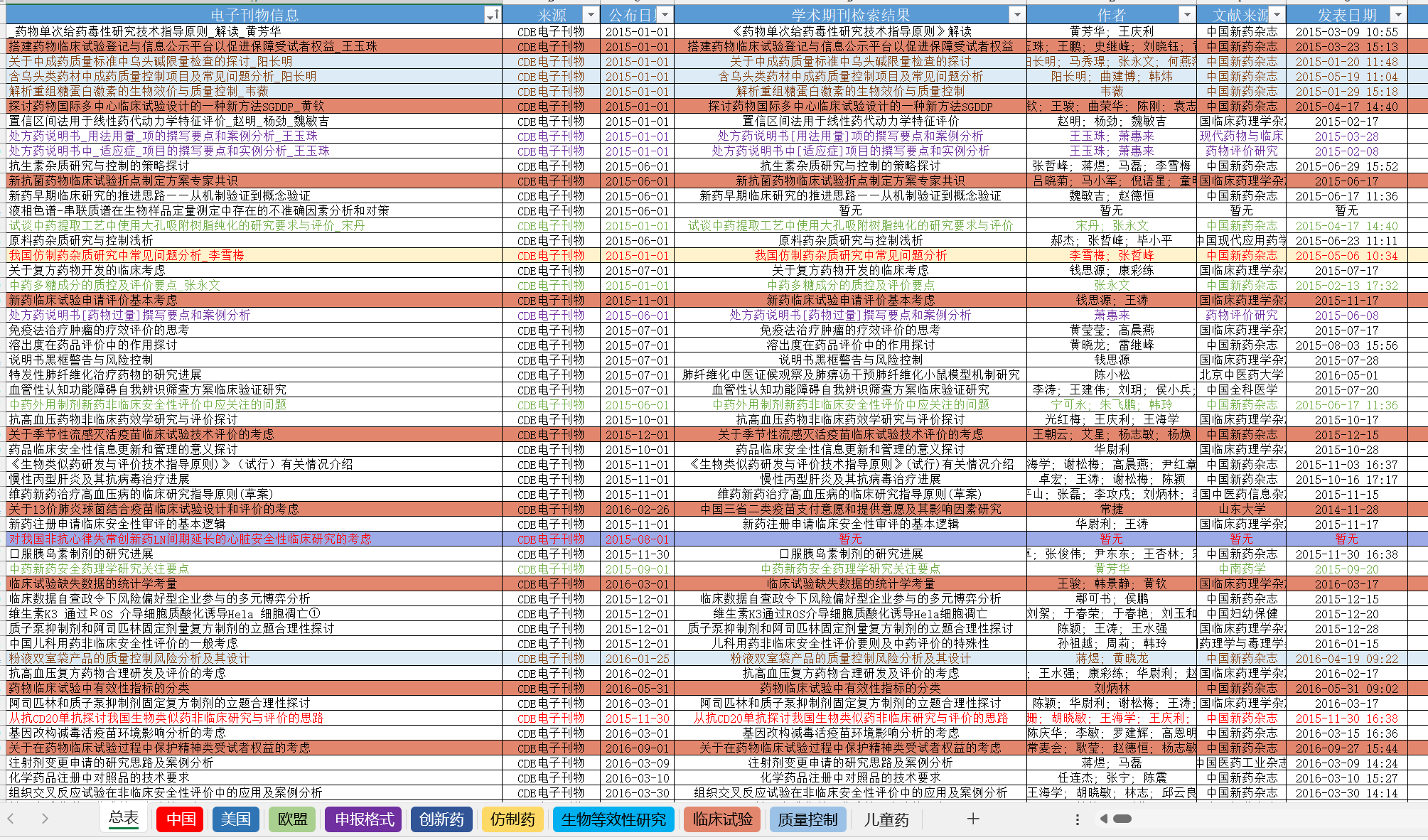
资源分享-CDE电子刊物总览
引言 在药品研发、审评和监管领域,及时获取专业、全面的信息至关重要。今天要给大家分享一份资源 ——CDE 电子刊物总览(2015-至今),这是一个涵盖丰富药品领域知识的信息宝库。 内...
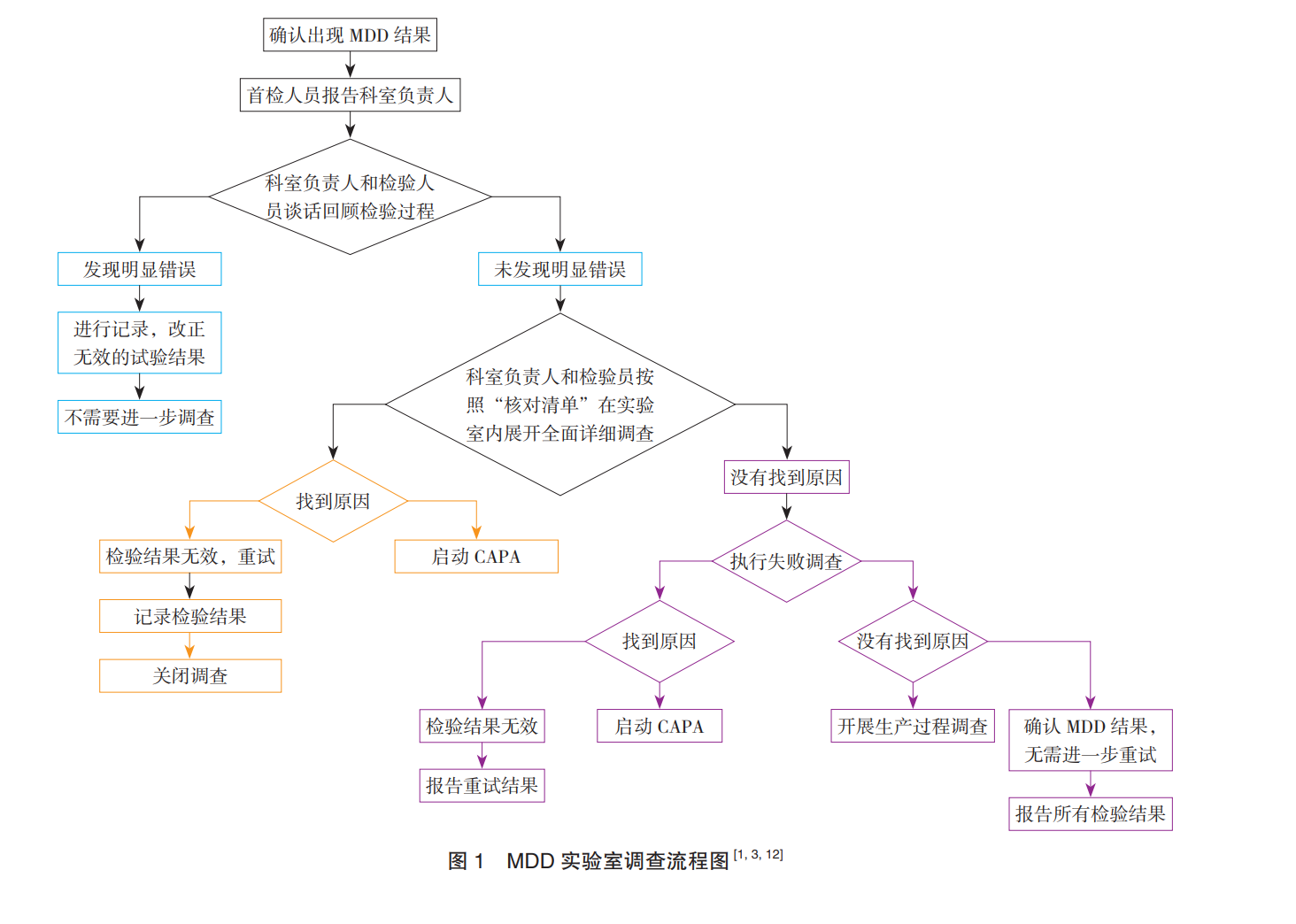
重磅文献分享-药品微生物数据偏差调查规范和研究概述-文末附下载地址
偏差是指使结果与事实之间存在差异的系统性倾向,数据的来源、数据处理方式等都可能导致偏差。微生物数据偏差(Microbial Data Deviation,MDD)于 2001 年由美国食品药品管理局(Food and ...

20211118-CDE-eCTD专题培训课件
2021年11月18日国家药品审评中心(CDE)主办的“中国eCTD进展情况概述”专题培训会的完整系列材料,共五份PPT,涵盖了eCTD从政策解读、技术规范、实施指南、申报流程到企业最佳实践的全链条内容...

202502-FDA-人工智能和机器学习在药品和生物制品研发过程中的运用Using Artificial Intelligence & Machine Learning in the Development of Drug & Biological Products-附下载
当人工智能(AI)与机器学习(ML)技术渗透到医疗健康的核心领域,药物开发这一长期被 “高投入、长周期、高风险” 困扰的行业,正迎来前所未有的变革契机。近日,美国食品药品监督管理局(FDA...





kidzhangy7月前0
感谢分享,楼主好人一生平安Terrific8月前0
感谢!特别有用,资源分享的很及时!北岛2年前0
相当于货架期多久?Mark4年前2
Thanks for your blog, nice to read. Do not stop.一位WordPress评论者5年前0
嗨,这是一条评论。 要开始审核、编辑及删除评论,请访问仪表盘的“评论”页面。 评论者头像来自Gravatar。