
行业资料共192篇 第3页
排序
分析天平的最小称量值可否通过增量法变相增加称样量以满足要求?药典2025给出答案
最近在论坛又看到有人在讨论最小称量值的问题,具体问题见下图。 其中一个小伙伴答复到 那么是否可以按照这位网友的评论进行操作呢?我实际在工作中遇到了类似问题,有的同事说他...
202512-广东药监-广东省药品审评检查沟通咨询问答500问-附下载
广东省药品审评检查沟通咨询问答500问,下载链接:https://pan.quark.cn/s/55172853f56c 第一篇 生物制品、麻醉药品、精神药品、医疗用毒性药品、药品类易制毒化学品、放射性药品...
20251203-EMA Classification of changes: questions and answers-EMA变更分类新规解析-附下载
欧盟药品管理局(EMA)的变更分类规则直接关系到产品上市后变更的合规推进。2025 年 11 月,EMA 更新了《变更分类问答手册》,针对行政变更、质量变更、(非)临床变更及编辑性变更四大核心场景...

202509-EMA-Guidelines on the details of the various categories of variation欧盟药品上市许可变更新指南(2026生效)-附下载
2026 年 1 月 15 日,欧盟《药品上市许可变更分类及流程指南》(C/2025/5045)将正式生效,替代沿用 13 年的 2013 版指南。这份经 2024 年修订的新规,通过简化流程、优化分类、强化工作共享机...
转载-2025年CDE官方问答全梳理(截至20260109)
2025年,国家药品监督管理局药品审评中心(CDE)持续围绕药品注册申报中的关键环节、高频疑问与共性难题,发布了一系列具有指导意义的共性问题解答。为帮助业界更清晰把握政策导向、提升申报效...

资源分享-药物研发基本原理 中文翻译版 第2版-本杰明-科学出版社
本书从药物发现与开发的历程开始, 为读者呈现了药物发现的经典靶点、体外筛选系统、 体内筛选模型、药物化学和药代动力学研究等新药研究的基本原理和方法, 同时介绍了药物临床前和临床研究过...

国内首个原料药连续制造指南-原料药及中间体连续制造指导原则(附下载)
今天分享一份重磅资料:《原料药及中间体连续制造指导原则》(意见征集稿),这是由中国化学制药工业协会(PIAC)发布的国内首个针对原料药连续制造的行业指南。旨在推动医药工业高端化、智能化...
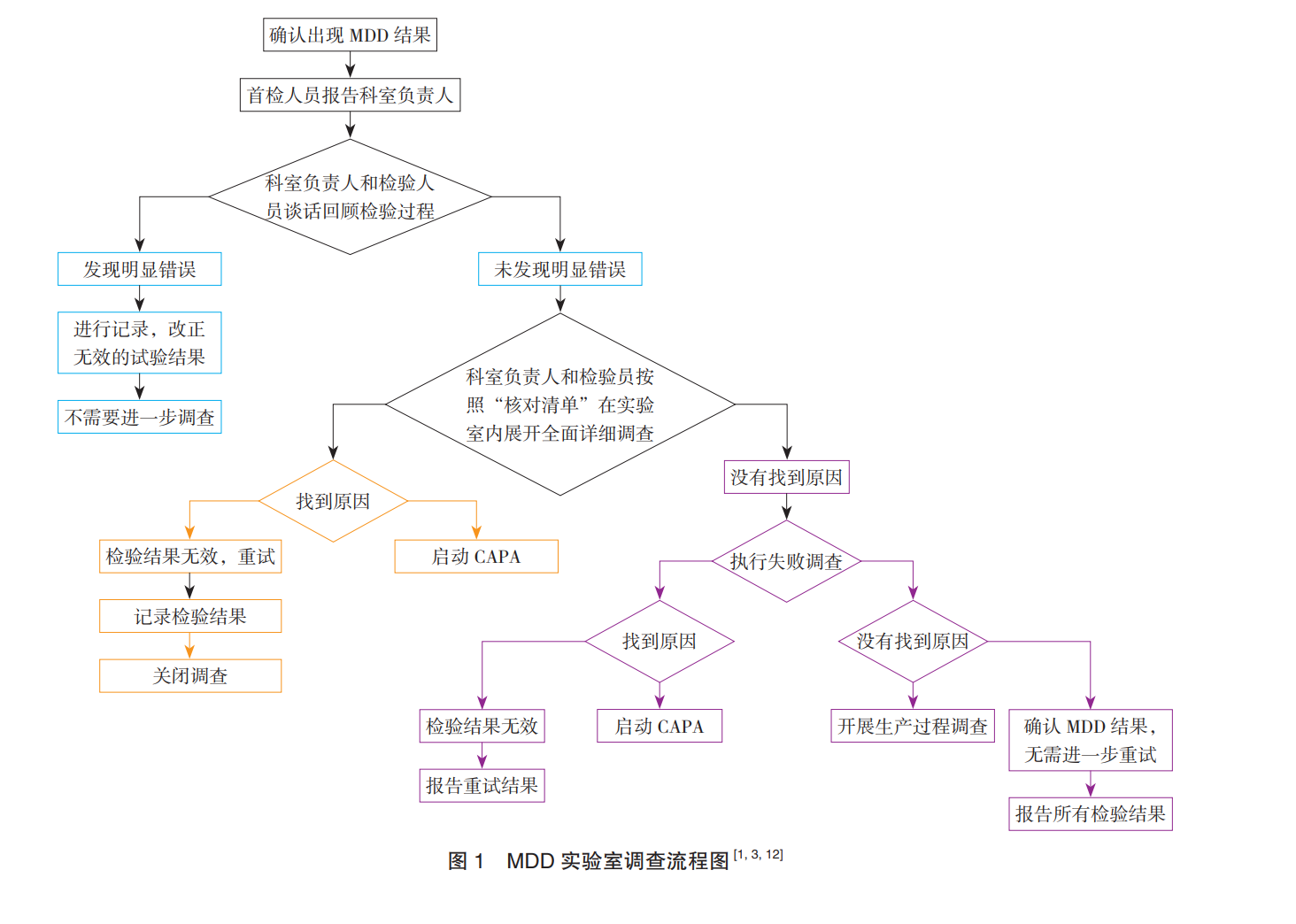
重磅文献分享-药品微生物数据偏差调查规范和研究概述-文末附下载地址
偏差是指使结果与事实之间存在差异的系统性倾向,数据的来源、数据处理方式等都可能导致偏差。微生物数据偏差(Microbial Data Deviation,MDD)于 2001 年由美国食品药品管理局(Food and ...

药审云课堂答疑汇总(2024年12月-2025年12月)-转自iReg
问题1:起始原料引入两个手性碳,可能存在4个异构体,并且起始原料无紫外吸收,异构体研究难度较大,不在起始原料中进行控制,在中间体及成品中研究是否可行? 解答1:首先建议参考ICH Q11及...
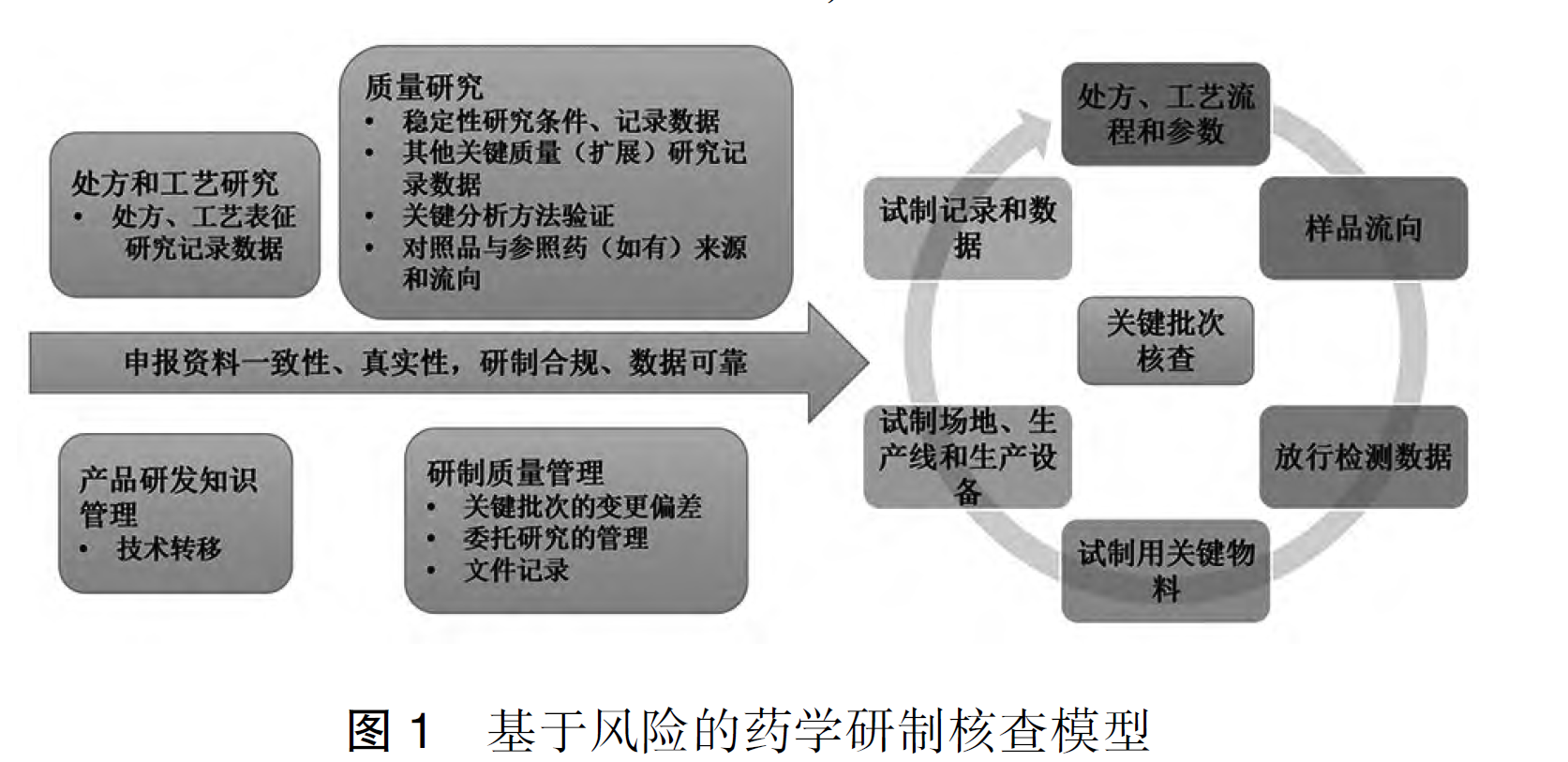
重磅文献分享-生物制品药学研制核查要点和常见问题分析(2025 CFDI 张平)-附下载
摘要 生物制品的生产和质量控制过程中,存在诸多可变性和特殊性。在新的药品注册监管法规体系下,研制现场核查是促进生物制品研发相关数据真实可追溯,推动生物制品研发行业规范和高质量发展...





kidzhangy7月前0
感谢分享,楼主好人一生平安Terrific8月前0
感谢!特别有用,资源分享的很及时!北岛2年前0
相当于货架期多久?Mark4年前2
Thanks for your blog, nice to read. Do not stop.一位WordPress评论者5年前0
嗨,这是一条评论。 要开始审核、编辑及删除评论,请访问仪表盘的“评论”页面。 评论者头像来自Gravatar。