
资源分享共115篇 第2页
排序

202509-EMA-Guidelines on the details of the various categories of variation欧盟药品上市许可变更新指南(2026生效)-附下载
2026 年 1 月 15 日,欧盟《药品上市许可变更分类及流程指南》(C/2025/5045)将正式生效,替代沿用 13 年的 2013 版指南。这份经 2024 年修订的新规,通过简化流程、优化分类、强化工作共享机...
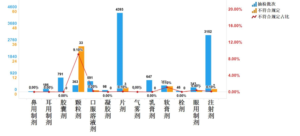
转载-2025年CDE官方问答全梳理(截至20260109)
2025年,国家药品监督管理局药品审评中心(CDE)持续围绕药品注册申报中的关键环节、高频疑问与共性难题,发布了一系列具有指导意义的共性问题解答。为帮助业界更清晰把握政策导向、提升申报效...
20251203-EMA Classification of changes: questions and answers-EMA变更分类新规解析-附下载
欧盟药品管理局(EMA)的变更分类规则直接关系到产品上市后变更的合规推进。2025 年 11 月,EMA 更新了《变更分类问答手册》,针对行政变更、质量变更、(非)临床变更及编辑性变更四大核心场景...
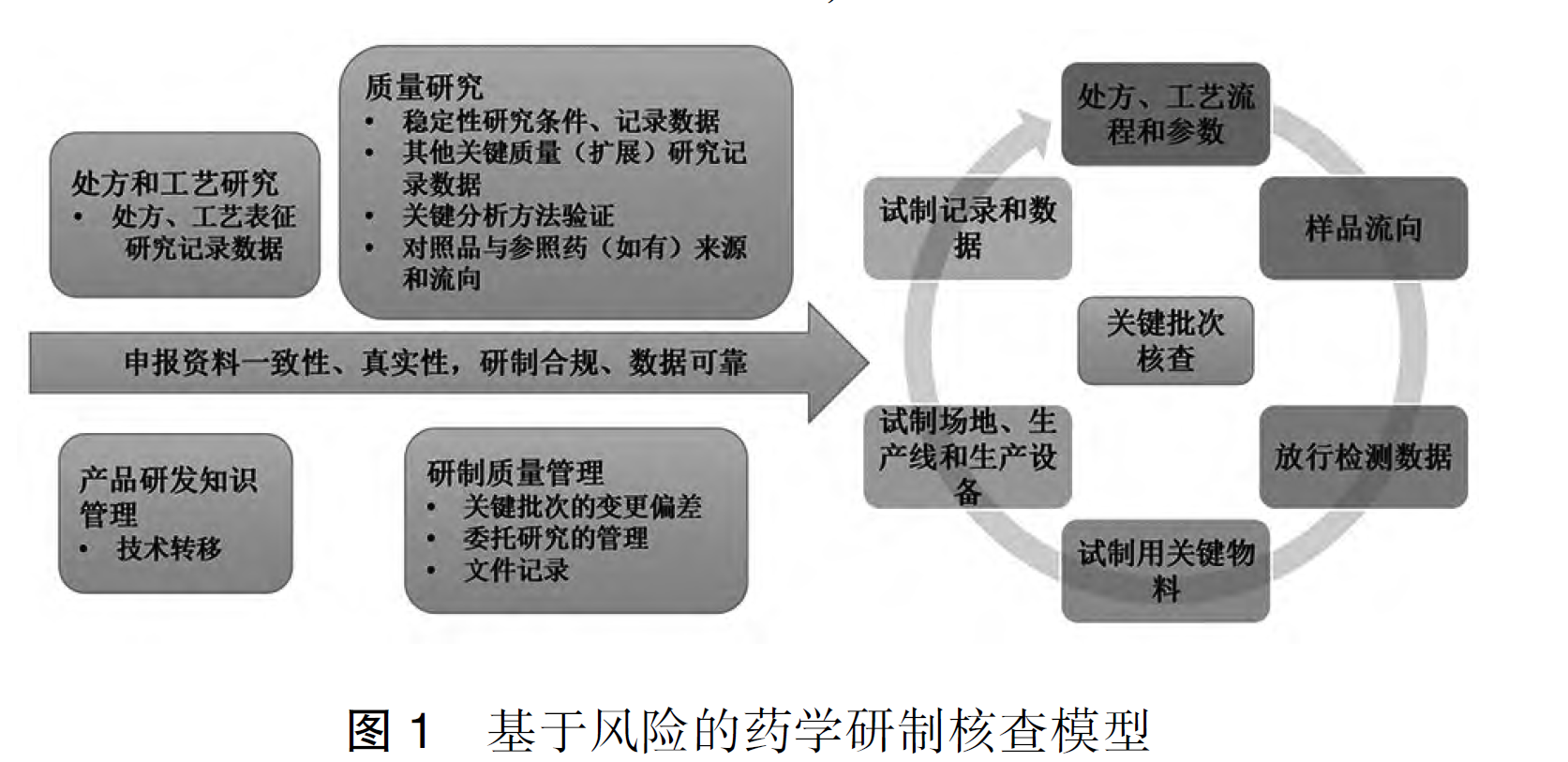
重磅文献分享-生物制品药学研制核查要点和常见问题分析(2025 CFDI 张平)-附下载
摘要 生物制品的生产和质量控制过程中,存在诸多可变性和特殊性。在新的药品注册监管法规体系下,研制现场核查是促进生物制品研发相关数据真实可追溯,推动生物制品研发行业规范和高质量发展...

资源分享-中检院-生物制品检验技术操作规范2019版
本书为《中国食品药品检验检测技术系列丛书》之一,其内容主要包括通用检测方法和各类生物制品的特异性检测方法。通用检测方法,只收录了至少两大类制品共同使用的方法,有些方法虽然在某一大类...

21 CFR Part 312 Investigational New Drug Application (IND)-新药临床试验申请 中英文双语对照
《21 CFR Part 312》是美国食品药品监督管理局(FDA)制定的关于新药临床试验申请 (IND)的管理法规,规定了药物在正式上市前开展临床研究的全过程监管要求。该法规最初于 1987 年发布,...

资源分享-202512-美国药典凡例(中英文对照)-允咨
该文件为美国药典(USP-NF)凡例的中英文对照版本,核心围绕药典的适用范围、关键规则及具体要求展开,明确其适用于 USP 和 NF 认可的所有专论与通则,规定了药典的标题版本(含常规修订与加速...
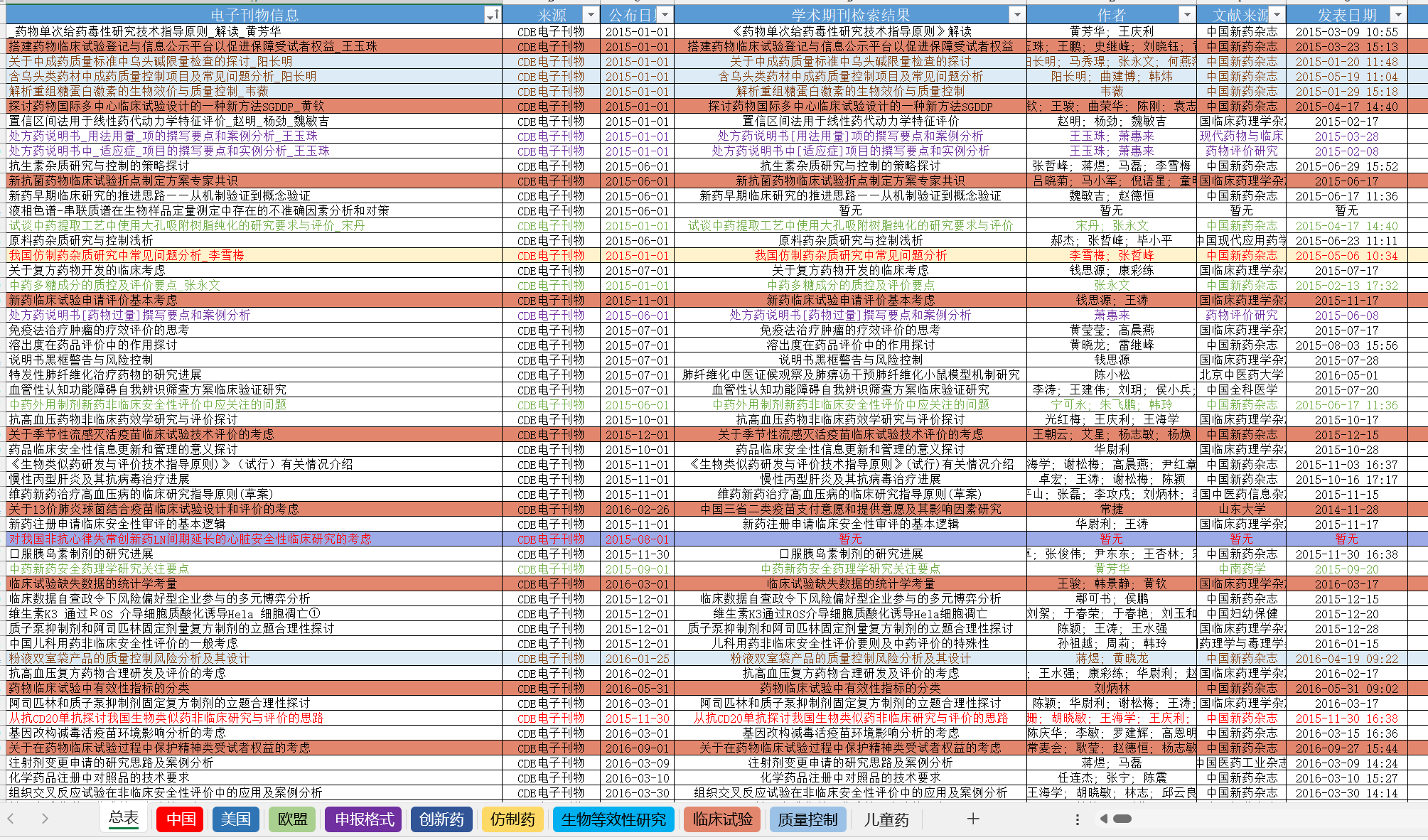
资源分享-CDE电子刊物总览
引言 在药品研发、审评和监管领域,及时获取专业、全面的信息至关重要。今天要给大家分享一份资源 ——CDE 电子刊物总览(2015-至今),这是一个涵盖丰富药品领域知识的信息宝库。 内...

资源分享-CMDE-中国医疗器械器审中心答疑解惑汇总(202511)
医疗器械器审中心发布的 6.3 版答疑解惑汇总(2025 年 11 月 17 日),收录了 2017 年至 2025 年间的 530 个核心问题及解答,聚焦医疗器械与体外诊断试剂注册申报、临床评价、检测验证、技术要...

书籍分享-GMP计算机化系统实施与检查指南(2025版张金贵)
《GMP 计算机化系统实施与检查指南》由张金贵所著,聚焦药品生产领域计算机化系统的 GMP 合规管理,系统涵盖计算机化系统的定义、分类、GMP 基本要求与管理框架,详细阐述系统开发与验证、运行...




kidzhangy8月前0
感谢分享,楼主好人一生平安Terrific9月前0
感谢!特别有用,资源分享的很及时!北岛2年前0
相当于货架期多久?Mark4年前2
Thanks for your blog, nice to read. Do not stop.一位WordPress评论者5年前0
嗨,这是一条评论。 要开始审核、编辑及删除评论,请访问仪表盘的“评论”页面。 评论者头像来自Gravatar。