
行业资料共192篇 第7页
排序
202512-广东药监-广东省药品审评检查沟通咨询问答500问-附下载
广东省药品审评检查沟通咨询问答500问,下载链接:https://pan.quark.cn/s/55172853f56c 第一篇 生物制品、麻醉药品、精神药品、医疗用毒性药品、药品类易制毒化学品、放射性药品...

资源分享-APIC 数据完整性问答 第三版 中英文对照(Data Integrity — Frequently Asked Questions (FAQ))
在 GMP/GxP 合规领域,数据完整性(Data Integrity) 一直是监管热点。无论是 FDA、EMA 还是中国 NMPA 的检查中,数据完整性问题频频被点名,稍有不慎就可能导致警告函或停产整改。今天,我为大...

资源分享-大湾区202509药品注册生产现场核查与符合性检查衔接工作程序介绍
对于药企而言,药品注册生产现场核查与上市前 GMP 符合性检查是药品上市前的关键环节。国家层面出台的衔接工作程序,不仅明确了核查检查的核心规则,更直接影响企业的注册效率与合规成本。今天...

202509-APIC-Quality Agreement Template for Regulatory Starting Materials("RSMs") and Critical Materials-APIC更新RSM关键起始物料质量协议模板-附下载
,原料药(API)的质量安全直接关乎终端药品的疗效与患者健康,而作为 API 生产关键环节的注册起始物料(RSMs)和关键物料,其质量管控离不开规范的合作协议支撑。近日,活性药物成分委员会(AP...

资源分享-20251205-江苏药监-江苏药品委托生产现场检查常见缺陷分析(南京分中心)
江苏省药品监督管理局审评核查南京分中心于 2025 年 12 月 5 日举办的药品委托生产现场检查常见缺陷分析培训,系统梳理了药品委托生产过程中在机构与人员、共线生产管理、委托生产管理等多个关...

资源分享-202512-EMA-Guideline on stability testing for applications for variations to a marketing authorisation(EMA 稳定性试验变更指南)
2025 年 12 月,欧洲药品管理局(EMA)发布了修订后的《上市许可变更申请稳定性试验指南》(EMA/CHMP/QWP/441071/2011-Rev.3),该指南将于 2026 年 1 月 15 日正式生效,全面替代 2011 年版 Re...

20251231-APIC-起始物料供应商质量协议模板Quality Agreement Template for Regulatory Starting Materials ("RSMs") and Critical Materials
这份文件是 2025 年 9 月版本的 APIC(活性药物成分委员会)质量协议模板,适用于两家公司之间注册起始物料(RSMs)及关键物料的合作,共包含引言 / 目的 / 范围、一般条款、质量责任等七大部分...

资源分享-CFDI20251015注册核查数据可靠性要求与常见问题分析-张平
在生物制品行业,数据可靠性是药品研发、生产与注册的 “生命线”。无论是疫苗、抗体药还是其他生物制品,其安全性、有效性和质量可控性都依赖全生命周期数据的真实、完整与可追溯。国家药监局...
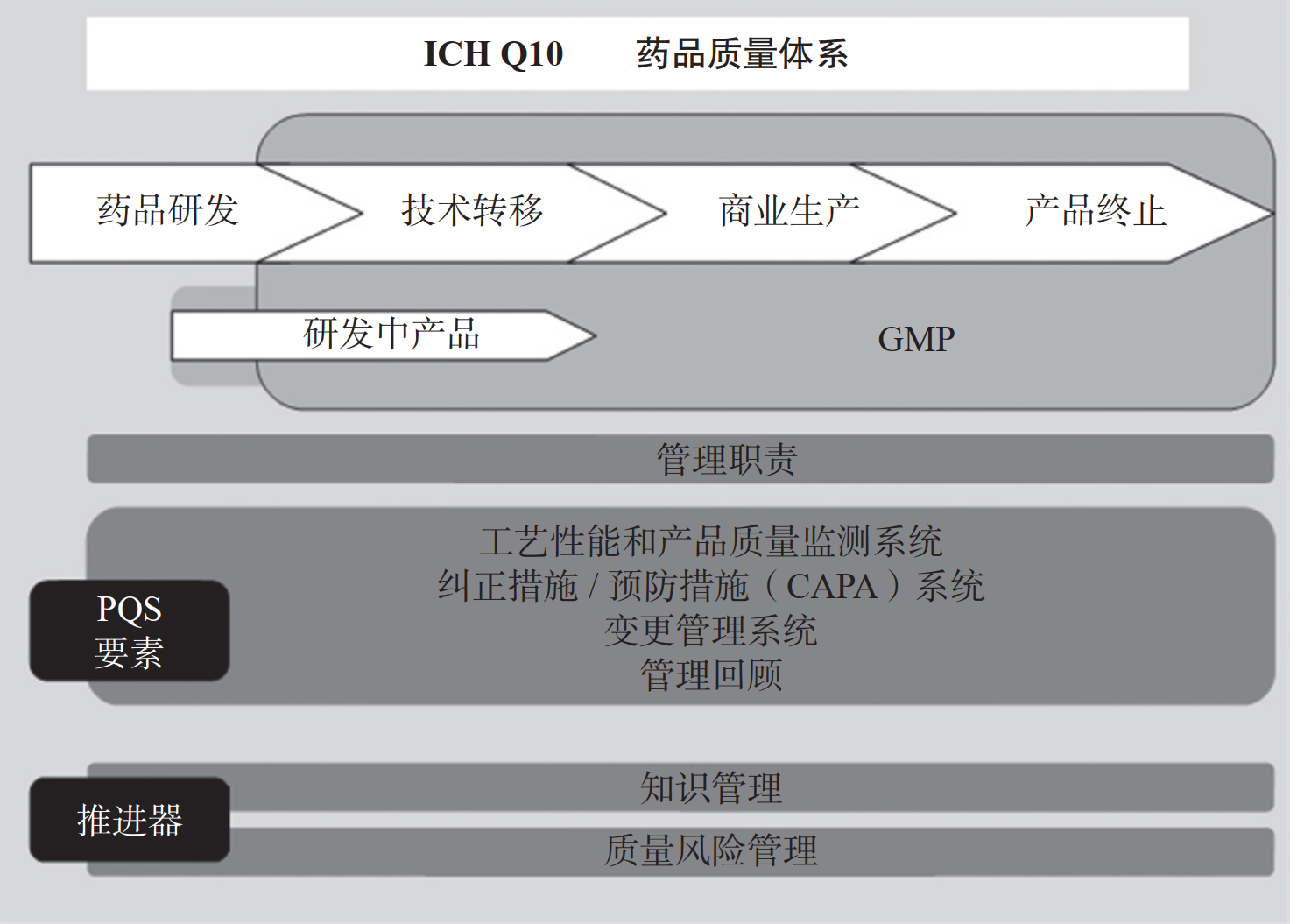
文献学习-MAH 制度下集团型企业一体化质量管理模式的实践与探索-附下载
摘要 在制药行业中,质量是企业生存和发展的基石,它不仅关系到企业的核心价值,更是赢得消费者信任和巩固市场竞争力的关键因素。当下制药企业面临的挑战日益复杂,需要不断创新发展并增强国...

资源分享-202512-美国药典凡例(中英文对照)-允咨
该文件为美国药典(USP-NF)凡例的中英文对照版本,核心围绕药典的适用范围、关键规则及具体要求展开,明确其适用于 USP 和 NF 认可的所有专论与通则,规定了药典的标题版本(含常规修订与加速...





kidzhangy8月前0
感谢分享,楼主好人一生平安Terrific9月前0
感谢!特别有用,资源分享的很及时!北岛2年前0
相当于货架期多久?Mark4年前2
Thanks for your blog, nice to read. Do not stop.一位WordPress评论者5年前0
嗨,这是一条评论。 要开始审核、编辑及删除评论,请访问仪表盘的“评论”页面。 评论者头像来自Gravatar。